Dr. Eugene Braunwald
University of California, San Diego, Hospital Practice,
The prolific sympathetic innervation of the heart permits synthesis of about 90% of the myocardial norepinephrine requirements within the organ. In congestive failure, the endocrine-like activity of the heart is almost totally eliminated. There is evidence that this deletion may deprive the failing heart of a normally available source for the augmentation of its contractility.
From the time that William Harvey discovered the prime function of the heart to be that of a pump, physiologists and clinicians have marveled at the remarkable ability of this organ to modify its performance according to the needs of the peripheral tissues. Radical alterations in heart rate and cardiac output are accomplished with ease, and in a matter of seconds. Efforts to understand better the mechanisms whereby the heart controls propulsion of blood through the circulatory system to meet changing conditions have occupied generations of researchers in the field of cardiovascular disease. Over the years this question has been explored at many levels: in isolated heart muscle, in whole heart, in heart-lung preparations, and in intact animals. More recently, refinement of investigative techniques has permitted more direct study of myocardial function in man.
A fact basic to regulation of myocardial function is that while heart muscle resembles other striated muscle structurally and biochemically, it differs from it in an important way. When called upon to increase contractile activity it does not do so by modifying the number of contractile units that play a role, as is the case with skeletal muscle. The magnitude of contractile response is controlled instead by the length of the muscle fibers at the time of activation, ventricular end-diastolic volume and filling pressure being the chief expressions of fiber length and tension.
But this control mechanism, despite its key role, is not the only one that regulates the heart’s performance. There is a second crucial mechanism, whereby changes in activity of the sympathetic nervous system can directly alter ventricular function at any given level of end-diastolic volume or pressure. Indeed, Starling himself, noted for elucidating the first intrinsic mechanism governing heart action, recognized the importance of neurohumoral adjustments in adapting ventricular function to the body’s requirements. As he observed in a paper published in 1920: “No understanding of the circulatory reactions of the body is possible unless we start first with the fundamental properties of the heart muscle itself and then find out how they are modified, protected, and controlled under the influence of mechanisms – nervous, chemical, and mechanical – which under normal conditions play upon the heart.”
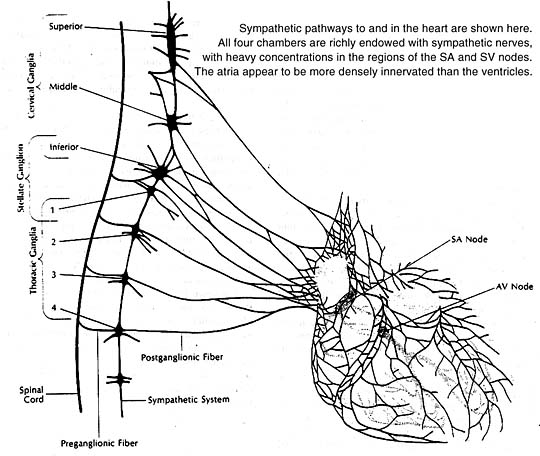
All four heart chambers are richly endowed with sympathetic nerves that serve as a link between the brain and contracting heart muscle. Nerve terminals storing the sympathetic neurotransmitter norepinephrine are found throughout the heart wherever their presence could count: in the sinoatrial and atrioventricular nodes, in the Purkinje system, in the atrial and ventricular myocardia. At these sites the neurotransmitter acts principally to stimulate the heart’s beta-adrenergic receptors, as can be confirmed experimentally by infusion of a beta-adrenergic stimulant such as isoproterenol. The responses elicited are similar to those produced naturally by norepinephrine release from sympathetic nerve endings.
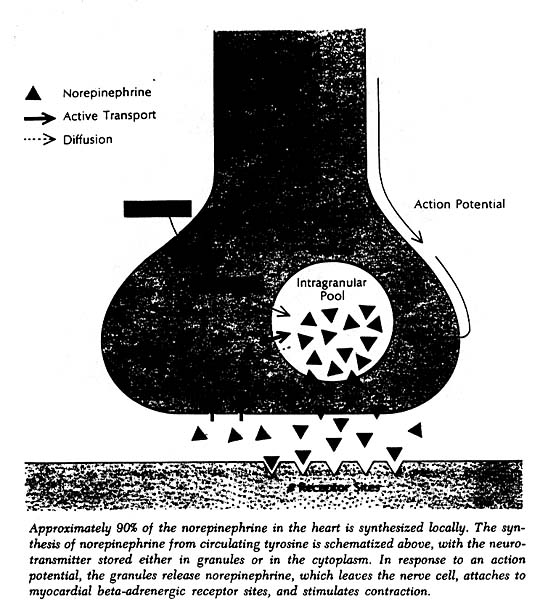
The heart, like other organs innervated by sympathetic nerves, can readily extract norepinephrine from the blood stream, and for a long time it was assumed that the heart’s norepinephrine stores were derived primarily from the blood. Within the past few years it has been demonstrated experimentally, both in canine and guinea pig heart preparations, that the heart itself and its constituent sympathetic nerve endings can synthesize the neurotransmitter from its precursor amino acid tyrosine and need not be dependent on extraction from the blood to maintain its stores. Indeed, it now appears that fully 90% of the norepinephrine present in the heart is manufactured there. In this sense one can think of the heart as an endocrine gland that synthesizes and releases a hormone, norepinephrine, as needed to allow the circulation to respond appropriately to changing metabolic demands of body tissues.
Norepinephrine acting on the sinoatrial node increases the rate of diastolic depolarization and thus speeds the heart rate. Norepinephrine acting on the atrioventricular node increases the velocity of conduction and diminishes the refractory period during which the AV node is unresponsive to stimuli coming from the atrium. By increasing the excitability of nonautomatic tissue, norepinephrine can also induce ventricular tachycardia and other arrhythmias. Most important of all from the point of view of normal cardiovascular function – as well as its maintenance in disease states – norepinephrine can significantly improve myocardial contractility.
Animal experiments a decade ago by Sarnoff and coworkers demonstrated that either electrical stellate ganglion stimulation or norepinephrine infusion augments the strength of atrial contraction and thus the atrial contribution to ventricular filling. Even more pertinent to the discussion to follow, they also showed that either nerve stimulation or neurotransmitter infusion markedly increases external stroke work and power produced by the ventricle at any given mean atrial pressure and at any end-diastolic pressure or fiber length.
The interest of our research group, first at the National Institutes of Health and more recently at the University of California, has been directed at assessing the importance of the sympathetic nervous system, particularly as it affects ventricular function, in normal circulatory regulation in man. A special concern also has been to assess the importance of the sympathetic nervous system in maintaining circulatory adequacy when the myocardium is depressed, or when an increased hemodynamic burden is placed on the heart because of an imbalance between cardiac output and the perfusion requirements of peripheral tissues.
An intriguing early experiment in our laboratories indicated the potential importance of the adrenergic nervous system in a man undergoing thoracotomy: Measurement of the effects of direct electrical stimulation of the sympathetic nerves with use of a myocardial strain gauge arch showed a consistent and significant increase in the force of ventricular contraction, although the length of segment of myocardium to which the strain gauge was sewn remained constant.
It seemed clear that stimulation of nervous pathways in the sympathetic nervous system could result in profound augmentation of the contractile state of the human myocardium. However, it could not be concluded from such stimulation experiments that the ordinary activity of the adrenergic nervous system has a significant effect on myocardial function in intact man.
Thus, several initial studies by our group were aimed at establishing normal parameters by evaluating the role of the sympathetic nervous system in exercise. Other investigators as well were suggesting that sympathetic activity could be important in mediating cardiovascular response to exercise, since many changes in cardiac performance during exercise resemble those resulting from stimulation of sympathetic pathways.
In one study we attempted to define the relationship of changes in heart rate, sympathetic activity, and ventricular dimensions in the cardiovascular response to mild exercise (performed on a bicycle ergometer in the supine position). Subjects were a group of patients being reevaluated some months after cardiac surgery. At the time of the exercise test they had only trivial abnormalities in cardiac function, so their responses might be considered analogous to those of normal subjects under similar conditions.
Dimensions of the left and/or right ventricle were obtained by measuring the distances between radiopaque markers (placed during cardiac surgery) on sequential frames of cineradiograms exposed at 30 frames per second; intraventricular pressure was simultaneously determined. The cineradiographic technique also permitted analysis of myocardial force-velocity relations, an analysis predicted on the fact that throughout active contraction the position of the force-velocity curve of the myocardium is determined by muscle length and contractile state. Thus, if the force-velocity relation is always examined at the same muscle length, this relation will define the contractile state of the myocardium.
The patients first performed light exercise as instructed; after a rest period, they repeated the exercise while the right atrium was electrically stimulated to keep the heart rate constant. The contribution of the sympathetic nervous system was assessed by administering the adrenergic-blocking agent propranolol before the exercise. (The availability of drug that selectively block activity of the adrenergic nervous system has made it far easier to estimate the role of this system in maintenance of myocardial function in man.)
During the first exercise period there was a decrease in ventricular end-diastolic dimensions; during exercise at a constant heart rate these dimensions increased. However, the most striking alteration was in the contractile state of the myocardium, as shown by the increased velocity of muscle fiber shortening regardless of intraventricular pressure and dimensions. For example, during the first exercise period – when cardiac response was allowed to occur spontaneously – velocity per isolength of muscle fiber increased by an average of 80%; at the same time cardiac output increased by only 36%. Thus the performance of the ventricle had improved, but clearly not as much as contractility.
The relative roles of both alterations in heart rate and sympathetic activity were also investigated with the subjects at rest but with induced tachycardia. The force-velocity relationship was shifted as before, but to a relatively small extent in comparison with that produced by exercise. Moreover, since greater shifts in force-velocity relationships occurred when exercise was performed at a constant rate, it would appear that the increase in heart rate in spontaneous exercise is a contributory but not the chief factor affecting the contractile state of the myocardium.
On the other hand, in the experiments with propranolol it was shown that adrenergic blockade almost completely prevented the exercise-induced shift in force-velocity relationships. The small residual increases in contractility that occurred with propranolol could be attributed to the elevation in heart rate that still occurred despite the block, since, when heart rate was also controlled, even these small shifts were abolished. Without adrenergic blockade, there was clearly an improvement during exercise in the contractile state of the myocardium, primarily because of increased sympathetic activity, which allowed the ventricle to eject blood more rapidly in order to meet increased metabolic requirements.
One interesting finding in these studies was that ventricular dimensions were reduced less during exercise than when a similar increase in heart rate was induced at rest. At any given heart rate, ventricular end-diastolic and end-systolic dimensions as well as stroke volume were all larger during exercise than at rest. Thus it can be seen that the Starling mechanism does in fact participate in the adaptation of the heart to exercise, even in the presence of an actively functioning sympathetic nervous system.
More information on the role of sympathetic activity in mediating cardiovascular response to exercise was sought in another study of healthy volunteers and patients with mild-to-moderate heart disease, who performed exercise on a motor-driven treadmill at varying speeds and inclines. Responses with and without adrenergic blockade with propranolol were determined at both submaximal and maximal exercise levels in healthy subjects and at submaximal levels in those with heart disease.
With adrenergic blockade, exercise at both levels invariably caused lesser increases in heart rate, cardiac output, mean arterial pressure, and left ventricular minute work. However, the fact that increases occurred despite adrenergic blockade confirmed that sympathetic nervous stimulation of the heart is not the only mechanism whereby heart rate and cardiac output are augmented during exercise.
In other experiments, sympathetic blockade produced with use of guanethidine again caused depression of the circulation during exercise. During a predrug exercise period, exertion resulting in a four-to-fivefold increase in oxygen consumption brought average increases above resting values of 68% in heart rate, 17% in stroke volume, 96% in cardiac output, and 129% in left ventricular minute work. After administration of both guanethidine and atropine, identical levels of exercise produced average increases above resting values of only 28% in heart rate, 1% in stroke volume, 30% in cardiac output, and 5% in left ventricular minute work. Blockade of the parasympathetic nervous system alone with use of atropine did not interfere with the circulatory response to exercise, whereas when only the sympathetic system was blocked with guanethidine, cardiac output, mean arterial pressure, and left ventricular work during exercise were significantly lower than during the predrug exercise period. Thus in the major contribution made by the autonomic nervous system in response to exercise it is the sympathetic division that clearly plays the critical role.
With these data in hand on the normal role of the sympathetic system in modulating cardiovascular function, the logical next step was to attempt to determine how the system might act to maintain myocardial function in patients whose cardiovascular reserve is reduced by failure. If for no other reason, the widespread clinical use of adrenergic-blocking drugs in such patients gave immediate practical importance to evaluation of sympathetic activity. We hoped these studies might also increase understanding of basic mechanisms that operate in heart failure.
A number of findings over the years had indicated that cardiovascular sympathetic nervous system function is altered in the clinical syndrome of congestive failure. The tachycardia, diaphoresis, peripheral vascular constriction, and suppression of urine formation in patients with heart failure had all been considered evidence of increased sympathetic nervous activity.
More direct evidence was obtained in a study in which subhypotensive doses of guanethidine were used to interfere with the activity of the sympathetic system in patients with heart failure. The 10 patients studied had inactive rheumatic valvular or primary myocardial disease. All had signs or symptoms of right- and/or left-sided congestive failure at the time of investigation. Manifestations of failure were increased in the five patients with more severe disease (functional classes III or IV by the New York Heart Association criteria). All showed an increase in dyspnea, orthopnea, and body weight; venous pressure and sodium excretion were also increased. In the five with less severe disease, congestive failure did not worsen with administration of the blocking agent.
Thus the sympathetic nervous system did appear to play a compensatory role in the circulatory adjustment of patients to heart failure. The findings also suggested a need for caution in the clinical use of adrenergic-blocking agents to treat patients with limited cardiac reserve.
More evidence for such a role was obtained by measurements of the concentration of norepinephrine in arterial blood. With sympathetic activity, a portion of the norepinephrine released from nerve terminals spills over into the bloodstream; determining the plasma norepinephrine concentration therefore provides a crude index to the level of sympathetic activity. Sensitive fluorometric techniques now available allow accurate measurement of norepinephrine levels not only in blood but in urine and tissue as well.
In normal subjects, moderate muscular exercise was associated with only a slight increase in arterial norepinephrine (from a mean of 0.28µ/L to 0.46µ/L). In patients with congestive heart failure, mean norepinephrine values were higher to begin with (0.63 µ/L) and increased to far higher values with exercise (to 1.73µ/L). Interestingly, resting and exercise levels in patients who had heart disease but were not in failure proved to be similar to those in normal subjects.
The elevated resting levels in patients with heart failure – and the excessive increase during exercise – evidently reflected an increased response of the sympathetic nervous system. The conviction grew that this increased response plays a useful supportive role in such patients.
Measurements of 24-hour urinary norepinephrine excretion were also made in a sizable group of subjects including 13 normal individuals, 30 patients with congestive failure, and 14 with heart disease but no failure. Norepinephrine excretion averaged 22µ/day in the normal subjects; it was significantly elevated in patients with heart failure, averaging 46µ/day in patients in functional class III and 58µ/day in patients in class IV. Again there was little or no difference between normal subjects and those with heart disease but no failure.
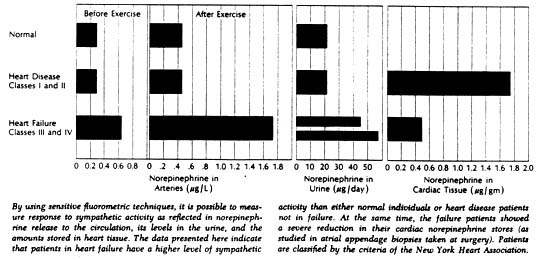
Given this evidence of hyperactivity of the sympathetic system in heart failure we wondered if cardiac stores of the neurotransmitter would be elevated. Accordingly, cardiac norepinephrine concentration was measured spectrofluorometrically in biopsy specimens of atrial tissue obtained from patients undergoing open-cardiac operations. The group included 34 patients who had not experienced failure prior to surgery and 49 who had. Unexpectedly, the norepinephrine concentration was significantly reduced in the patients with heart failure; it averaged 0.49µ/gm of tissue in patients who had been in failure as against 1.77µ/gm in those who had not. Extremely low values were fond in some patients with failure – norepinephrine concentrations of less than 10% of the average normal level.
Concentrations of the neurotransmitter were also measured in papillary muscles removed from the left ventricles of patients undergoing mitral valve replacement; these levels were markedly depressed in patients with mitral regurgitation who had been in severe left ventricular failure. Comparison of norepinephrine concentrations in ventricular and atrial tissues in individual patients showed a significant positive correlation.
The observed reduction in cardiac norepinephrine concentration might have been sufficient to impair adrenergic function, provided it was true depletion. Conceivably, the amount of norepinephrine per unit weight of tissue could be reduced merely because the presence of hypertrophied myocardium or fibrous tissue diluted a fixed number of normal nerve endings. This was one of several questions most readily studied in animal models of heart failure. Three mammalian species with chronic heart failure produced by different interventions were included so as to approximate, if possible, the several forms of heart failure encountered clinically. In the guinea pig, primary left heart failure was induced by constriction of the ascending aorta; in the dog, primary right heart failure was induced by surgery to create pulmonary stenosis and tricuspid insufficiency; in the cat, right ventricular hypertrophy with and without heart failure was induced by different degrees of pulmonary artery constriction.
In all three animal species, heart failure was associated with profound depletion in cardiac norepinephrine concentration. The depletion was evident in both right and left ventricles no matter which ventricle carried the primary hemodynamic burden. In the experimental animal, the total content of cardiac norepinephrine (as well as its concentration) can be readily determined; this value was also markedly depressed in animals with heart failure. In dogs with heart failure, reduction of the norepinephrine stores in the heart was sufficient to abolish the contractile response of isolated papillary muscle to the pressor amine tyramine, which acts by causing release of norepinephrine. In other words, true depletion of cardiac norepinephrine appears to occur in failure rather than the dilution of a normal complement by enlargement of the heart.

The time required after onset of heart failure for depletion of neurotransmitter stores was determined in the guinea pig. The concentration was found to be normal one day after aortic constriction but by the fifth day it had decreased to 22% of normal values. By that time substantial ventricular hypertrophy had already occurred. Values remained depressed for the 65 days during which the experiment was continued. Norepinephrine stores were also measured in guinea pig kidney to determine whether the depletion involved adrenergic nerves in other organs. There was no consistent change in renal stores for the failure group as a whole, providing further evidence that the norepinephrine depletion was limited to cardiac tissue.
The next task was to attempt to define the mechanism by which the depletion occurs. Defects of synthesis, uptake, and binding of norepinephrine or increased neurotransmitter turnover could all have been potential causes, and thee possibilities were all investigated. From the evidence it appeared that a serious defect in both norepinephrine synthesis and neuronal binding exists in the failing heart.
The defect in binding of norepinephrine was observed by measuring the norepinephrine retained in the harts and kidneys of guinea pigs after infusion of l-norepinephrine. In normal animals, ventricular and renal concentrations rose to peak values at the completion of the infusion; the concentrations declined over the ensuing three hours to values approaching control levels. In contrast, in animals with heart failure the same dose of neurotransmitter produced smaller increments in norepinephrine concentration and lower peak values. When tracer quantities of tritium-labeled dl-norepinephrine were injected, similar results were obtained. An hour later the left ventricles of the normal guinea pigs contained twice as much of the labeled norepinephrine as did those with heart failure.
It was reasoned that the altered capacity to retain administered neurotransmitter might be due to either a reduction in the total number of nerves in the myocardium or a diminution in the number of intraneuronal binding sites, or both. The subcellular distribution of administered radioactive norepinephrine was evaluated in the NE-depleted failing dog heart; the similarity of distribution in the normal and the failing heart pointed to a reduction in the number of nerve endings.
Net turnover rates were measured by administering a tracer dose of radioactive norepinephrine to guinea pigs and following the specific activity for 72 hours. In both normal and failing animals the decline in specific activity was complex and exhibited two exponential components (compatible with the occurrence of a multi-compartmental distribution of norepinephrine, as suggested also by other investigators). Absolute levels of specific activity and rates of norepinephrine disappearance were essentially the same in normal animals and in those with heart failure, indicating similar net turnover rates. But if smaller increments in norepinephrine levels after infusion in the failing animals could not be explained in terms of more rapid net turnover of neurotransmitter, this more strongly suggested that an abnormality of uptake and/or binding was at least partly responsible for the depletion. In view of the smaller norepinephrine stores, the presence of normal net turnover rates also suggested that the rate of formation might actually be reduced.
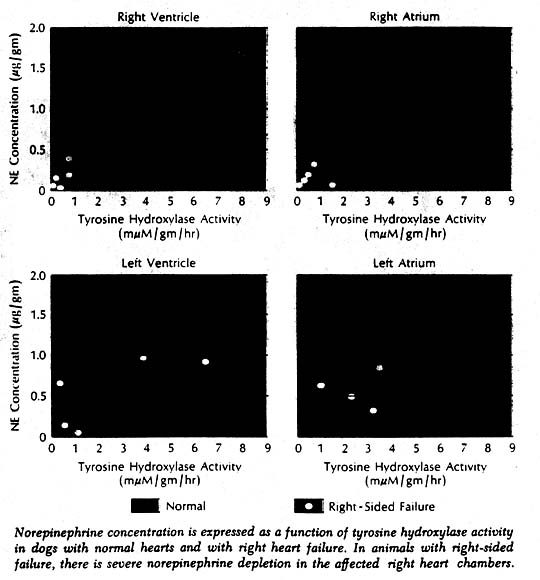
More direct evidence of a defect in neurotransmitter synthesis was obtained in enzyme studies in dogs with induced right heart failure and severe cardiac norepinephrine depletion. It has been known for some years that biosynthesis of norepinephrine proceeds through a series of steps from tyrosine to dopa to the immediate precursor dopamine. More recently it was learned that tyrosine hydroxylase, which catalyzes the first of these reactions (tyrosine to dopa), is the rate-limiting enzyme in synthesis of the neurotransmitter. Tyrosine hydroxylase activity in the right ventricle was markedly reduced in the affected dogs. Furthermore, there was a highly significant positive correlation when norepinephrine concentration and enzyme activity were related to each other in individual chambers of normal and failing hearts. Thus it appeared highly likely that the reduction of enzyme concentration was responsible for the cardiac neurotransmitter depletion in heart failure. What caused the reduced enzyme activity and depleted norepinephrine stores remains a puzzle.
The biochemical abnormality did not seem to lay a primary role. It seemed more likely that the reduced enzyme activity and depletion of norepinephrine occurred secondarily, perhaps as a result of a prolonged intensive barrage of sympathetic activity serving to bolster activity of the failing heart. Among findings suggesting that this is so is the fact that patients in whom overall activity of the sympathetic nervous system seems most intense, as reflected in elevated blood and urinary norepinephrine levels, have shown the most striking reductions in neurotransmitter stores. However, at present the mechanism ultimately responsible for the reduction in enzyme activity and norepinephrine synthesis remains to be elucidated. What can be said about the effects of the profound depletion of cardiac neurotransmitter on heart function? Could this be the basis for the intrinsic weakness of failing heart muscle?
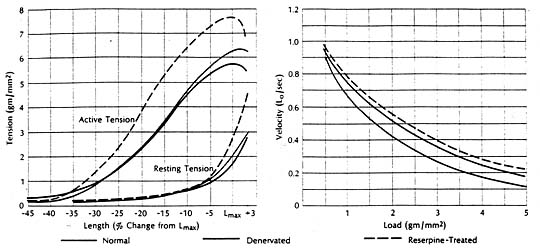
It had been shown experimentally in cats that congestive heart failure is associated with marked depression of the contractile state of cardiac muscle, reflected in a downward shift of the force-velocity curve and a reduction in the speed of contraction. The ventricles from which these failing muscles were removed had been fond depleted of cardiac norepinephrine. But the question remained whether the cardiac norepinephrine depletion as such was responsible for the depression of contractile performance in the failing heart. To find the answer it was necessary to examine the contractile state of cardiac muscle depleted of norepinephrine by a mechanism other than heart failure. The method selected entailed total chronic extrinsic denervation by means of mediastinal neural ablation (an operative procedure developed by Dr. Theodore Cooper); the cat papillary muscle was selected since extrinsic cardiac denervation had already proved feasible in this species. These experiments were also aided by newer methods for extending quantitative analysis of the mechanical properties of skeletal muscle to isolated preparations of myocardium.
Findings were compared in muscles obtained from normal animals and from those depleted of norepinephrine by cardiac denervation or by treatment with reserpine. The contractile state of the norepinephrine-depleted myocardium proved well within normal limits, whether depletion was achieved pharmacologically or with use of cardiac denervation. There was little difference in the three groups in terms of resting and active length-tension curves or force-velocity relations, or in augmentation of isometric tension achieved by paired electrical stimulation or by increasing the frequency of contraction in stepwise increments. It seemed evident then that the cardiac norepinephrine depletion that occurs in congestive heart failure is not responsible for the depression of failing heart muscle. Apparently cardiac stores of norepinephrine are not fundamental to maintaining the intrinsic contractile state of the myocardium. However, other findings indicate that cardiac norepinephrine depletion may seriously impair adrenergic function by interfering with release and transmission of neurotransmitter. For example, in one pertinent study the norepinephrine concentration in heart muscle was related to tis ability to augment tension on stimulation with the norepinephrine-releasing sympathetic amine tyramine. The muscles wee obtained from patients undergoing cardiac surgery. There was a significant correlation between norepinephrine concentration and maximum tension. Only muscles with higher concentrations could increase active tension further in response to tyramine, indicting impaired release in muscles having significantly reduced stores of neurotransmitter.

Another pertinent finding is that norepinephrine-depleted muscle from the failing heart tends to be supersensitive to exogenous norepinephrine. When the inotropic effect of l-norepinephrine was compared in heart muscle from normal cats and cats with pulmonary artery constriction, all muscles responded to exogenous norepinephrine with an increase in tension. But increments were actually somewhat greater in muscles from failing hearts, suggesting an increased responsiveness to the positive inotropic effects of norepinephrine. This observation, coupled with the knowledge that arterial norepinephrine concentration is markedly increased in heart failure, strengthens the view that the circulating catecholamines may for a time play an important role in maintaining the contractile function of the failing heart. However, with progressive heart failure and increasing dependence on sympathetic activity the circulating catecholamines may no longer be able to provide sufficient support. Gradually, as the cardiac norepinephrine stores become seriously depleted, there develops a relative deficiency of sympathetic function, which in turn adversely affects contractile function. If at this point a drug such as a beta-adrenergic receptor blocker that prevents circulating catecholamine from stimulating the myocardium is administered, the patient’s condition may be aggravated rather than improved.
The suggestion that there may be significant interference with transmission of sympathetic impulses as heart failure advances is based on experiments in dogs subjected to graded electrical stimulation of sympathetic nerves. Chronotropic and inotropic responses were compared in normal dogs and in dogs with right heart failure and cardiac norepinephrine depletion. Increments in heart rate and in ventricular contractile force were sharply reduced in the dogs with heart failure. In these same experiments it was shown that the heart muscle of dogs with heart failure continued to respond to exogenously administered norepinephrine.
Thus, while the norepinephrine depletion of congestive failure does not impair cardiac performance by altering the basic contractile state of the heart, it may indeed impair it by interfering with the augmentation in contractility that normally is readily provided by the sympathetic nervous system. From studies to date a key defect seems to be a drastic decrease in the quantity of neurotransmitter released per nerve impulse. The reduction in maximal response to sympathetic stimulation seen in animals with experimental failure suggests that even an abnormal increase in the impulse traffic along the cardiac sympathetic nerves does not substantially augment the contractile state of the myocardium. In this way, a critical mechanism for improving contractile force of the failing heart may go awry, unfortunately at the very time when it is sorely needed to prevent intensification of the heart failure.
